
Mengen-Treemaps
In den letzten Jahren etablierte sich die Quantitative Biologie in den Biowissenschaften. Besonders die Systembiologie verlangt nach quantitativen Daten, um zelluläre Prozesse mathematisch zu modellieren. Dies beeinflusst die Arbeit in unserem Haus bis heute. Zahlreiche Experimentalmethoden zur quantitativen Erfassung von Proteinen (Moleküle pro Zelle) wurden entwickelt oder für unsere Experimente angepasst. Auch hier war es wichtig, diese Entwicklung durch entsprechende Visualisierungsmethoden zu flankieren. Unter Umständen sollte man wissen, wieviel der ihr zur Verfügung stehenden Ressourcen eine lebende Zelle in die Synthese bestimmter Proteine investiert, um sich mit einem Umweltstimulus auseinanderzusetzen. Bei Hitze kann beispielsweise beobachtet werden, dass bei Bakterien 4% der zellulären Ressourcen nur dafür aufgewendet werden, um das Protein GroEL zu produzieren.
GroEL hilft bei der korrekten Faltung von Proteinen. Für Durchflussanalysen von Stoffwechselwegen ist es wichtig, die Mengen der entsprechenden Proteine (z.B. Enolase in der Glykolyse) zu kennen.
Da sich in lebenden Zellen mehrere Tausend Proteinarten befinden, können Kreis- oder Säulendiagramme eine entsprechende Visualisierung der Daten nicht mehr leisten. Darum entwickelten wir mathematische Verfahren, die iterativ die Fläche der Treemap-Zellen so anpassen, bis sie die Menge der Proteinmoleküle widerspiegeln. Mehrere Tausend Proteinmengen können so dargestellt, direkt verglichen und ihren Funktionskategorien zugeordnet werden.

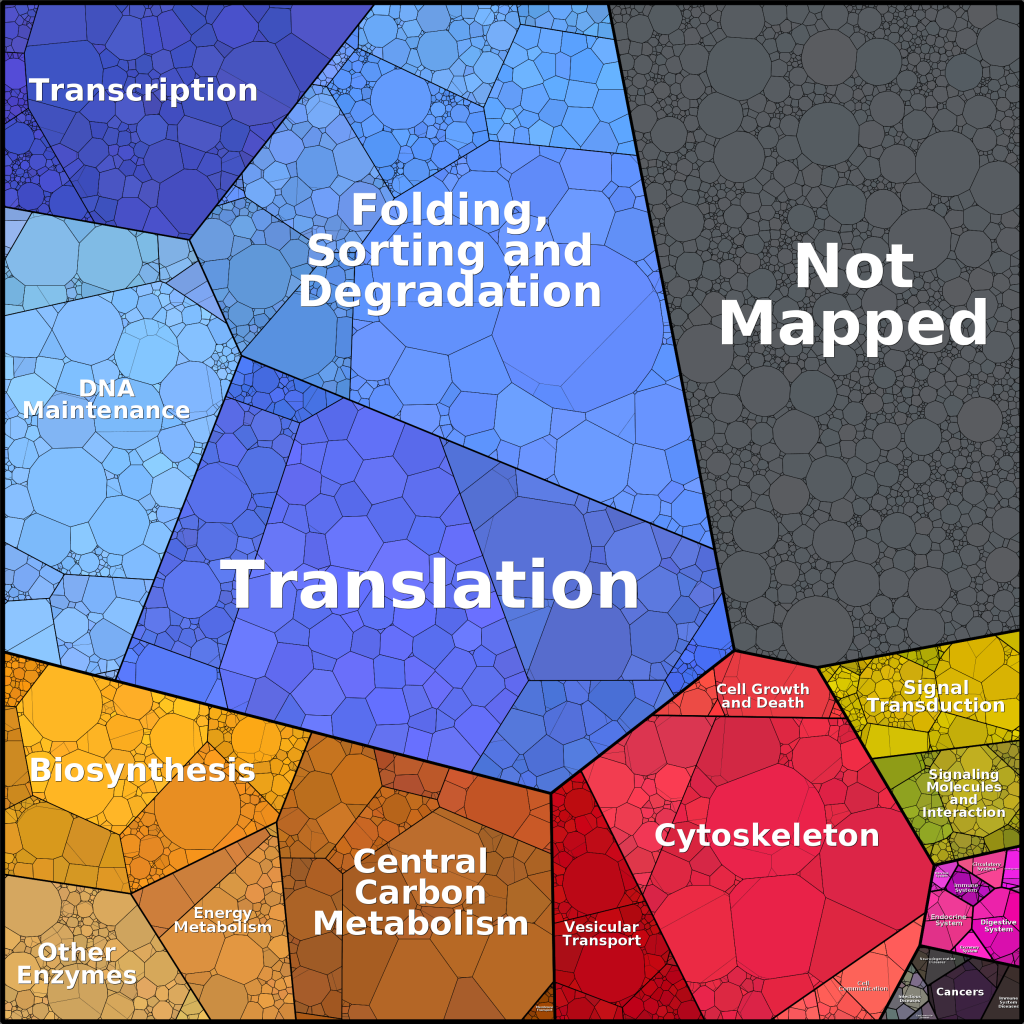
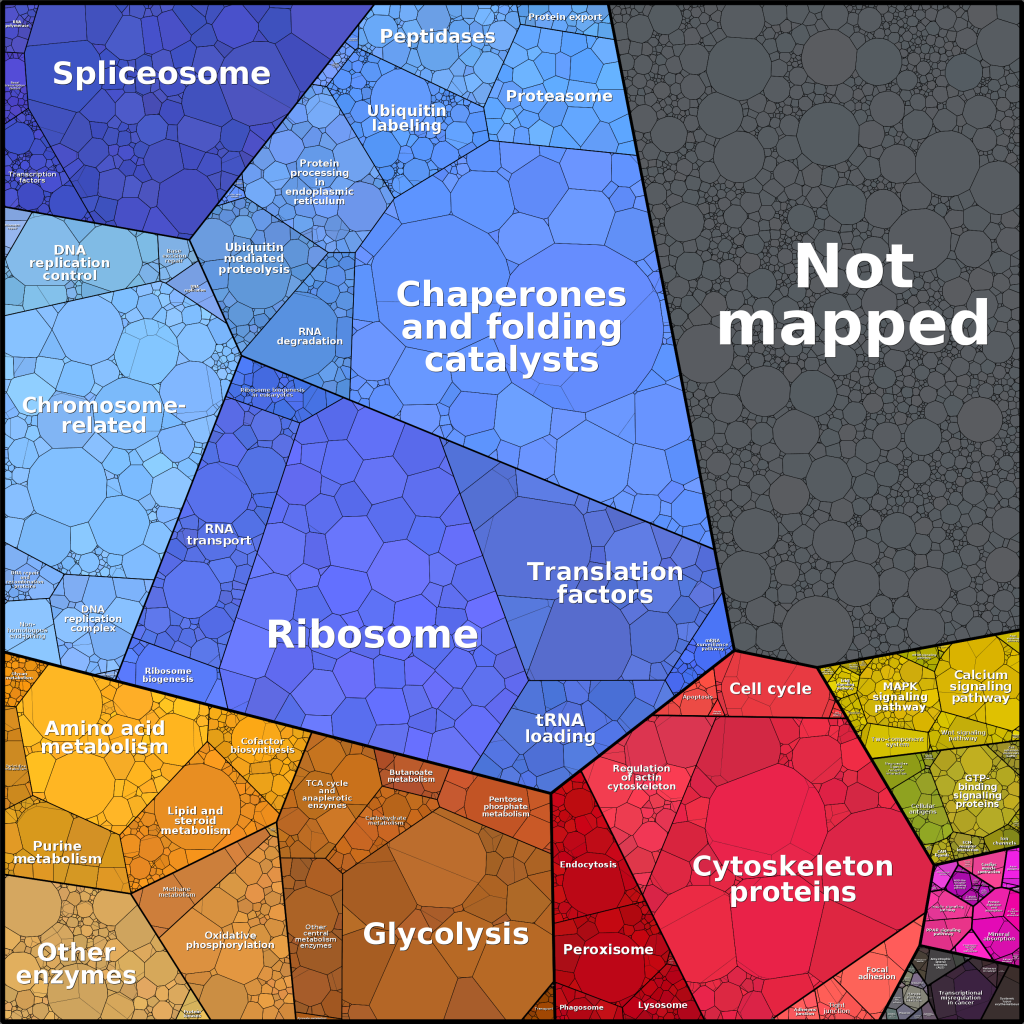
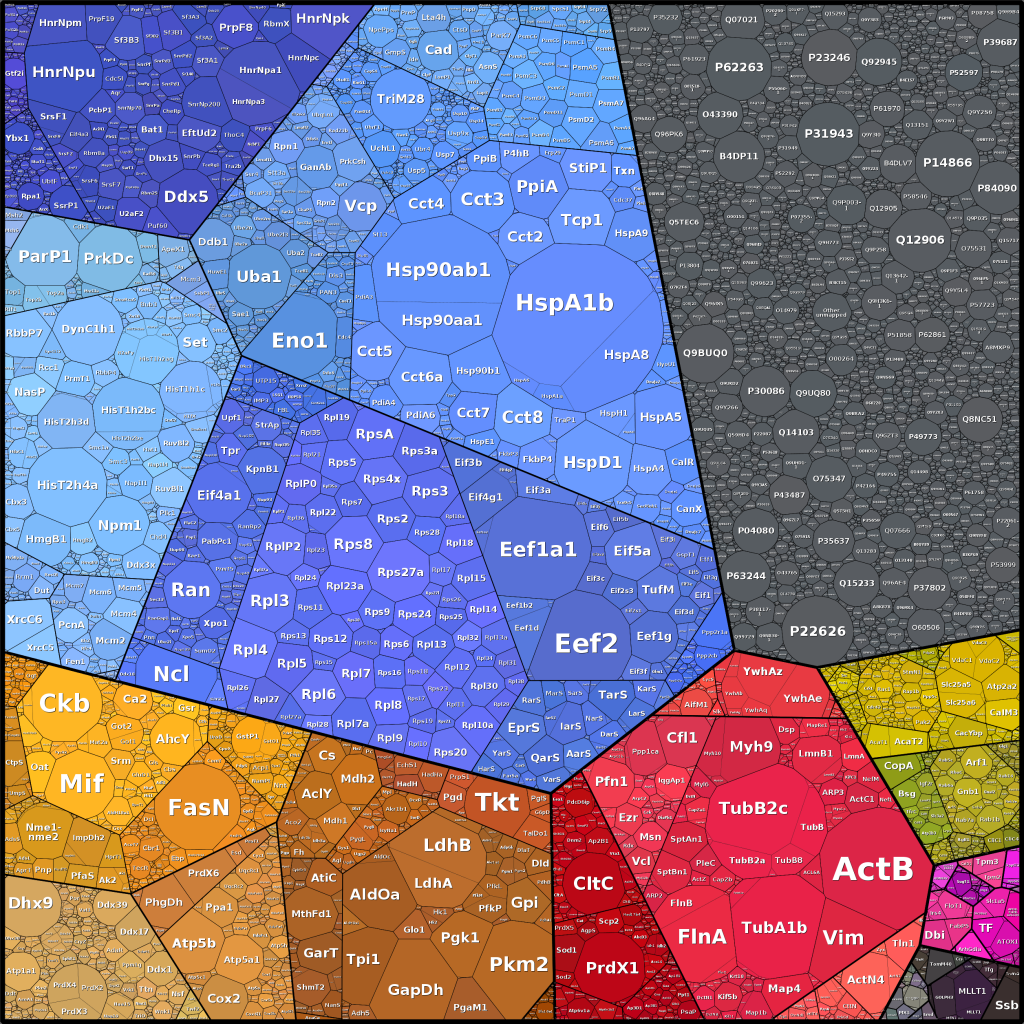
Die Einzelbilder zeigen die verschiedenen Stufen der Funktionshierarchie einer Krebszelllinie (HEK) entsprechend der Funktionszuordnung der Kyoto Encyclopedy of Genes and Genomes (KEGG). Die Farben visualisieren die Funktionshauptkategorien, die Zellflächen die Proteinmengen.
Erstmalig zeigten wir diese Art der Darstellung auf der Visualisierungskonferenz VizBi2014. In Zusammenarbeit mit Ron Milo vom Israelischen Weizmann Institut etablierten wir den Website www.proteomaps.org zur Präsentation quantitativer Proteomdatensätze und zur Erstellung eigener Mengentreemaps. Auch für Meta-Proteom-Studien können die gezeigten Mengentreemaps eingesetzt werden.